5. CpG_distrb_gene_centered.py¶
5.1. Description¶
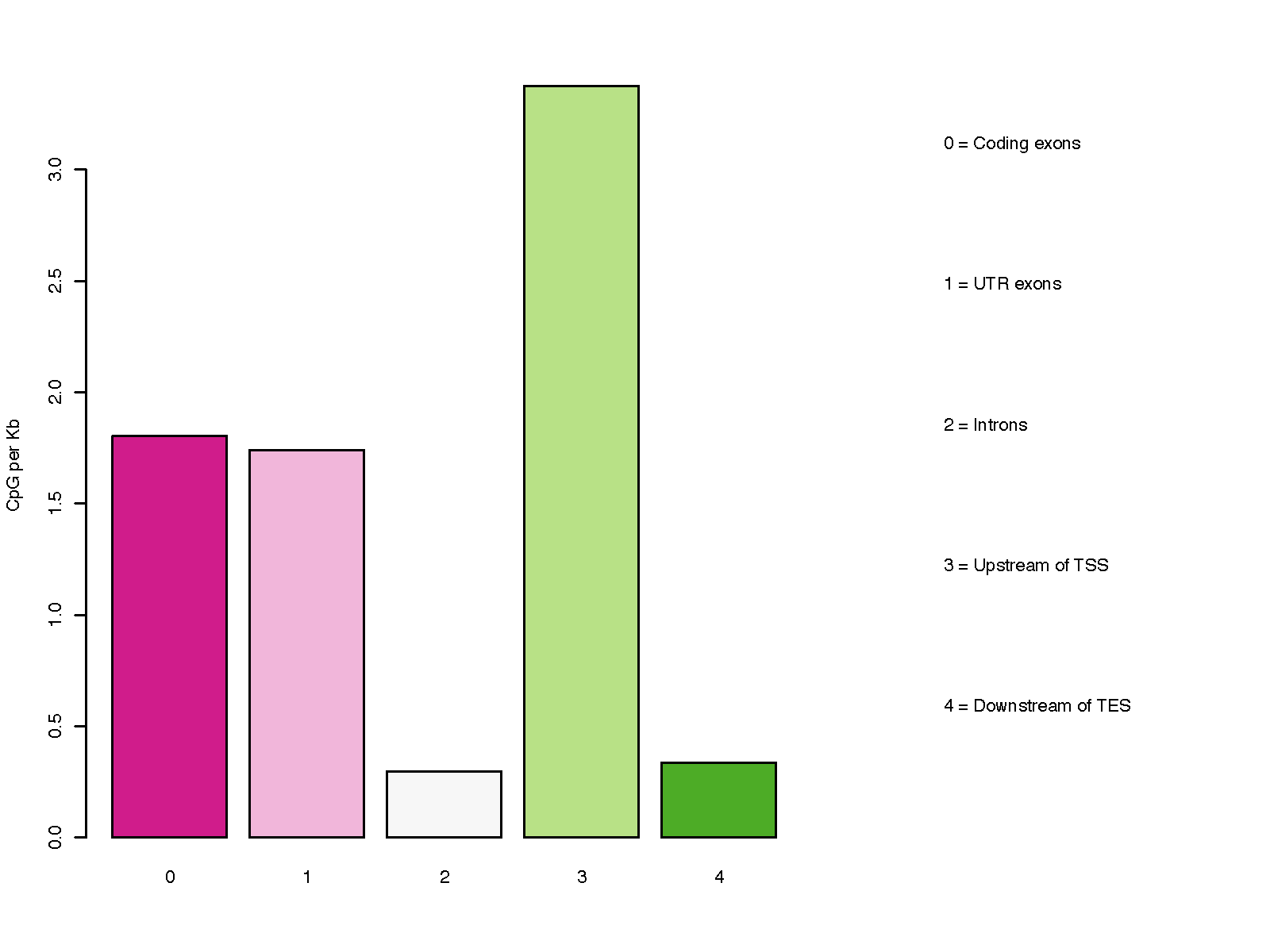
This program calculates the distribution of CpG over gene-centered genomic regions including ‘Coding exons’, ‘UTR exons’, ‘Introns’, ‘ Upstream intergenic regions’, and ‘Downsteam intergenic regions’.
Notes
Please note, a particular genomic region can be assigned to different groups listed above, because most genes have multiple transcripts, and different genes could overlap on the genome. For example, an exon of gene A could be located in an intron of gene B. To address this issue, we define the priority order as below:
Coding exons
UTR exons
Introns
Upstream intergenic regions
Downstream intergenic regions
Higher-priority group override the low-priority group. For example, if a certain part of an intron is overlapped with an exon of other transcripts/genes, the overlapped part will be considered as exon (i.e., removed from intron) since “exon” has higher priority.
5.2. Options¶
- --version
show program’s version number and exit
- -h, --help
show this help message and exit
- -i INPUT_FILE, --input_file=INPUT_FILE
BED file specifying the C position. This BED file should have at least three columns (Chrom, ChromStart, ChromeEnd). Note: the first base in a chromosome is numbered 0. This file can be a regular text file or compressed file (.gz, .bz2).
- -r GENE_FILE, --refgene=GENE_FILE
Reference gene model in standard BED-12 format (https://genome.ucsc.edu/FAQ/FAQformat.html#format1).
- -d DOWNSTREAM_SIZE, --downstream=DOWNSTREAM_SIZE
Size of down-stream intergenic region w.r.t. TES (transcription end site). default=2000 (bp)
- -u UPSTREAM_SIZE, --upstream=UPSTREAM_SIZE
Size of up-stream intergenic region w.r.t. TSS (transcription start site). default=2000 (bp)
- -o OUT_FILE, --output=OUT_FILE
The prefix of the output file.
5.3. Input files (examples)¶
5.4. Command¶
$ CpG_distrb_gene_centered.py -i 850K_probe.hg19.bed3.gz -r hg19.RefSeq.union.bed.gz -o geneDist