12. beta_profile_gene_centered.py¶
12.1. Description¶
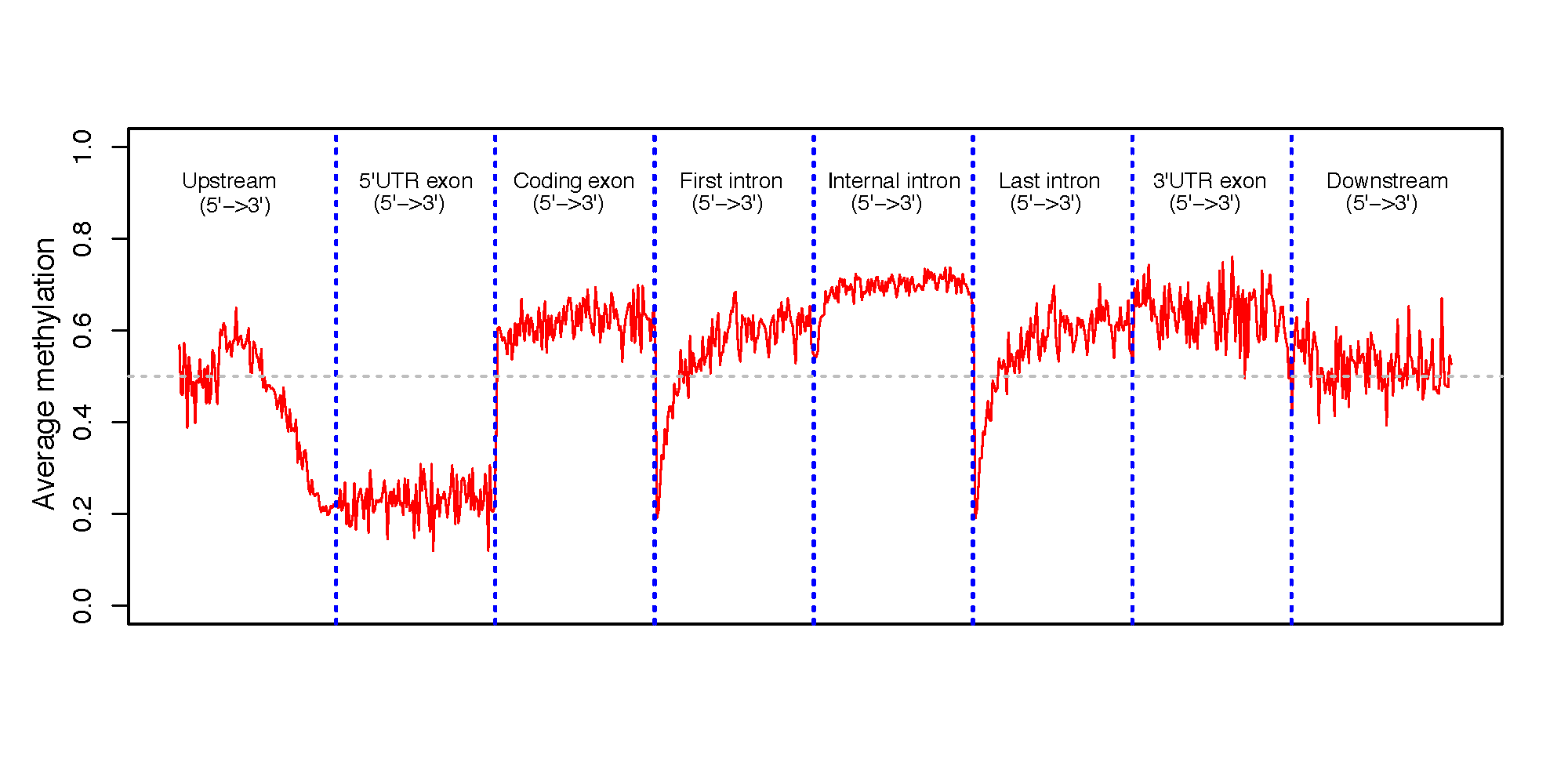
This program calculates the methylation profile (i.e., average beta value) for genomic regions around genes. These genomic regions include:
5’UTR exon
CDS exon
3’UTR exon,
first intron
internal intron
last intron
up-stream intergenic
down-stream intergenic
Example of input (BED6+)
chr22 44021512 44021513 cg24055475 0.9231 -
chr13 111568382 111568383 cg06540715 0.1071 +
chr20 44033594 44033595 cg21482942 0.6122 -
12.2. Options¶
- --version
show program’s version number and exit
- -h, --help
show this help message and exit
- -i INPUT_FILE, --input_file=INPUT_FILE
BED6+ file specifying the C position. This BED file should have at least 6 columns (Chrom, ChromStart, ChromeEnd, Name, Beta_value, Strand). BED6+ file can be a regular text file or compressed file (.gz, .bz2).
- -r GENE_FILE, --refgene=GENE_FILE
Reference gene model in standard BED12 format (https://genome.ucsc.edu/FAQ/FAQformat.html#format1). “Strand” column must exist in order to decide 5’ and 3’ UTRs, up- and down-stream intergenic regions.
- -d DOWNSTREAM_SIZE, --downstream=DOWNSTREAM_SIZE
Size of down-stream genomic region added to gene. default=2000 (bp)
- -u UPSTREAM_SIZE, --upstream=UPSTREAM_SIZE
Size of up-stream genomic region added to gene. default=2000 (bp)
- -o OUT_FILE, --output=OUT_FILE
The prefix of the output file.
12.3. Command¶
$beta_profile_gene_centered.py -i test_02.bed6.gz -r hg19.RefSeq.union.bed.gz -o gene_profile